

Um sich Molekülstrukturen besser vorstellen zu können, nutzen Sie entweder einen Molekülbaukasten oder greifen zur Visualisierung auf einen 3D-Moleküleditor wie Avogadro 2 zurück.
Nicht immer steht direkt ein Molekülbaukasten griffbereit, der es ermöglicht, ein Molekül kurzerhand aus allen Winkeln zu betrachten. Außerdem können Moleküle aus zig Atomen bestehen, sodass es deutlich einfacher ist, sie am Computer zu visualisieren. An dieser Stelle ersetzt der Moleküleditor Avogadro 2 [1] den Baukasten und erstellt anhand von SMILES-Strings komplexe Verbindungen. Um die Software richtig zu nutzen, sollten Sie einerseits Experimentierfreude mitbringen und andererseits einschlägige Literatur wie das E-Book “Learning Avogadro: The Molecular Editor” [2] zu Hilfe nehmen.
Hinter Avogadro 2 verbirgt sich ein plattformübergreifendes C++-Programm, das sich als Linux-Variante bereits in den Repositories der gängigen Distributionen findet. Unter Ubuntu richten Sie das Programm beispielsweise mit dem Befehl aus Listing 1 ein. Alternativ gibt es auch ein entsprechendes Appimage [3].
Listing 1
Installation
$ sudo apt-get install avogadro avogadro-utils molequeue
Moleküle zeichnen
Unter Avogadro 2 empfiehlt es sich, Moleküle anhand von SMILES- oder InChI-Strings zu erzeugen. Solche Repräsentationen von chemischen Strukturen als Zeichenketten finden Sie auf diversen Webseiten, zum Beispiel PubChem [4]. Unter Struktur | Einfügen geben Sie die gültigen String-Repräsentationen der Moleküle ein und lassen per Klick auf OK das Molekül bauen.
Darüber hinaus bringt das Tool eine Bibliothek mit Vorlagen zu etlichen Molekülen mit, sodass Sie diese nicht selbst zeichnen müssen. Die Library findet sich unter Struktur | Einfügen | Fragment. Sofern das gewünschte Molekül weder in den Vorlagen noch als gültige String-Repräsentation im Internet existiert, nutzen Sie ein Programm wie JChemPaint [5] oder eine Online-App wie Molinspiration [6] zum Zeichnen und generieren damit einen gültigen SMILES-String.
Finden Sie die Suche nach der passenden String-Repräsentation einer chemischen Struktur zu aufwendig, können Sie versuchen, in der Alltagssprache gebräuchliche Namen einzugeben. Zur Namensauflösung verbindet sich Avogadro 2 mit einem Server [7], der die dazugehörige chemische Struktur ermittelt. Die Suche selbst starten Sie unter Datei | Importieren | Download by name. Allerdings funktionierte im Test lediglich die Eingabe von “water”; andere korrekte umgangssprachliche Namen bekannter, chemischer Strukturen wie “acetic acid” lieferten kein Ergebnis.
Mit Avogadro 2 lassen sich Moleküle alternativ auch per Hand zeichnen, indem Sie auf das Stift-Icon klicken und links das entsprechende Atom sowie die nötige Bindungsart auswählen. Anschließend fügen Sie das Atom per Linksklick ins Zeichenfeld ein. Setzen Sie beim Zeichnen ein Häkchen bei Wasserstoffatome adjustieren, passt das Tool die Wasserstoffatome nach jeder Änderung automatisch an. Zum Ersetzen eines Atoms durch ein anderes genügt es, auf der linken Seite eines auszuwählen und mit der linken Maustaste auf das zu ersetzende Wasserstoffatom zu klicken (Abbildung 1). Zudem gibt es unter Struktur | Wasserstoffatome weitere Funktionen, die sich mit Wasserstoffatomen beschäftigen. Beispielsweise können Sie hier zusätzliche Wasserstoffatome hinzufügen oder entfernen.
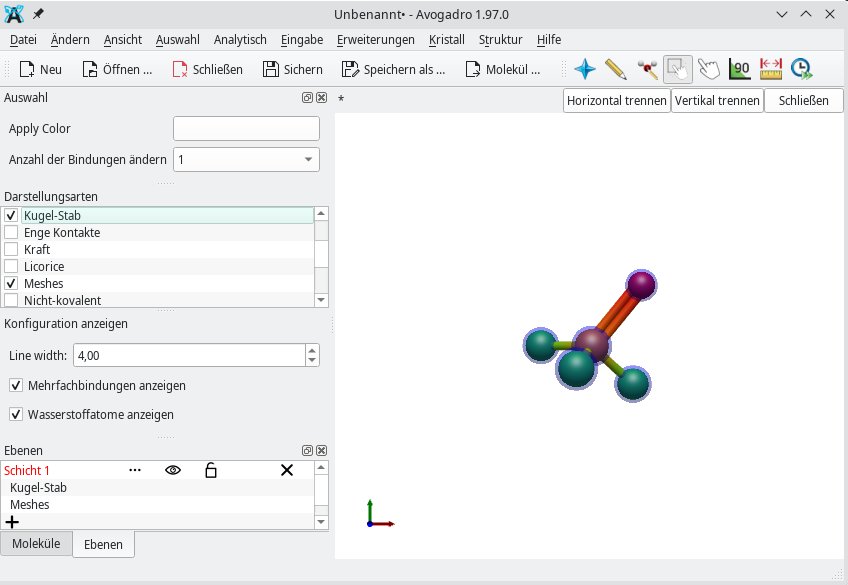
Abbildung 1: Manuell gezeichnete Moleküle (hier Phosphoroxychlorid, POCl3) gelingen nicht immer auf Anhieb.
Moleküle visualisieren
Die Visualisierung eines Moleküls lässt sich in Avogadro 2 auf der linken Seite des Fensters konfigurieren. Für dreidimensionale Grafiken gibt es gleich mehrere geeignete Darstellungsformen. Bei Ringen wie Chlorpyridin können Sie beispielsweise sowohl Meshes (Netze) als auch Beschriftung und Kugel-Stab verwenden. Dadurch kennzeichnen Sie die einzelnen Atome oder nummerieren Sie durch, was sich vor allem bei organischen Verbindungen als nützlich erweist.
Zusätzlich verfügen die einzelnen Darstellungsarten über weitere Optionen, die das Programm einblendet, sobald Sie auf die entsprechende Darstellungsart klicken. Unter Beschriftung ändern Sie zum Beispiel die Hintergrundfarbe. Kugel-Stab ermöglicht, die Größe der Kugeln sowie Stäbe zu skalieren. Die Kombination aus Kugel-Stab, Van der Waals und Meshes sorgt für einen raumfüllenden Effekt, der insbesondere die Wirkung der Londonschen Dispersionskraft verdeutlicht.
Eine andere Form der Van-der-Waals-Kraft sind die Dipol-Dipol-Wechselwirkungen. Mithilfe der Darstellungsarten Van der Waals sowie Van der Waals (AO) lassen sich Dipolmomente am besten veranschaulichen (Abbildung 2). Anders verhält es sich mit der Darstellungsart Drahtgitter: Sie bildet Bindungen als Linien ab, sodass die Form einer Linienzeichnung ähnelt (Abbildung 3).
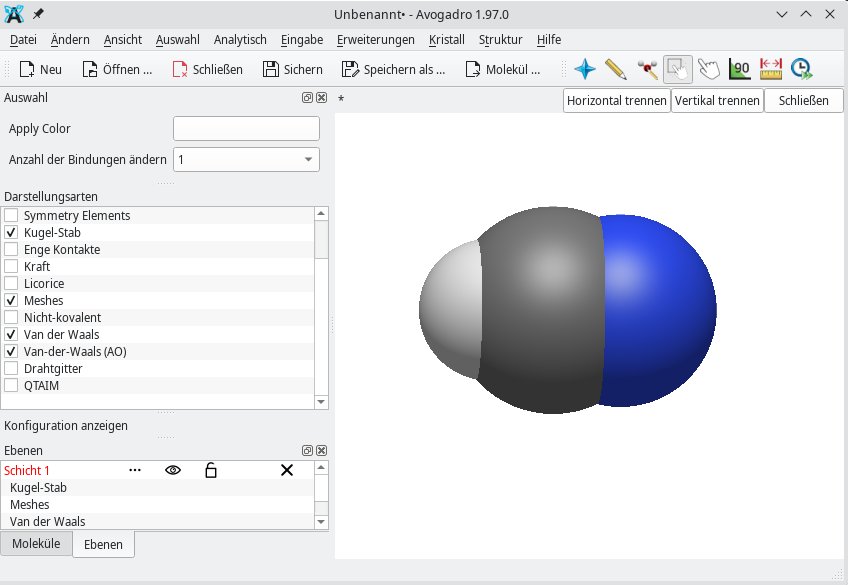
Abbildung 2: Anhand der Größe der Van-der-Waals-Kräfte lässt sich die Richtung des Dipolmoments erahnen. Im Beispiel CHN zeigt der Dipolvektor eindeutig in Richtung des Stickstoffatoms.
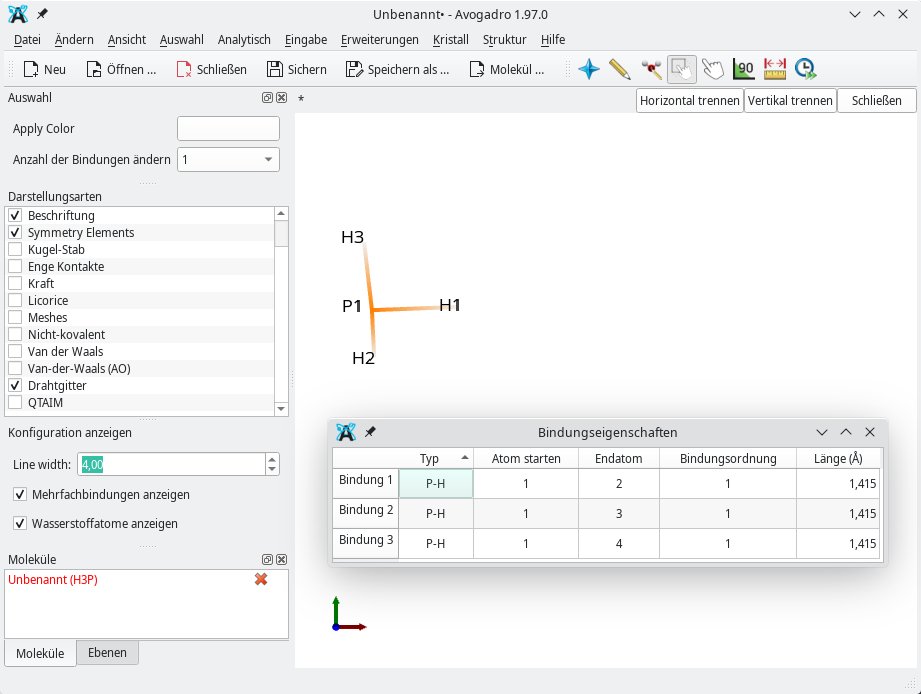
Abbildung 3: Die Darstellungsart Drahtgitter bildet den Phosphorwasserstoff (Monophosphan, PH3) als Linienzeichnung ab.
Außerdem können Sie anstelle der Kugel-Stab-Darstellung auf Licorice (Lakritz) umsteigen. Diese Darstellung unterscheidet zwar nicht zwischen den einzelnen Bindungsarten wie Einfach- oder Doppelbindung, hebt aber die Geometrie des Moleküls hervor. Dementsprechend eignet sie sich vor allem zum Drehen der Atome. Zuerst markieren Sie das gesamte Molekül mittels des Auswahlwerkzeugs (Zeigefinger-auf-Rechteck-Icon). Durch Aktivieren des Manipulationswerkzeugs (Zeigefinger-Icon) drehen Sie anschließend bei gedrückter linker Maustaste das Molekül um die horizontale Achse. Bei gedrückter rechter Maustaste hingegen lässt sich das Molekül in alle Richtungen drehen (Abbildung 4).
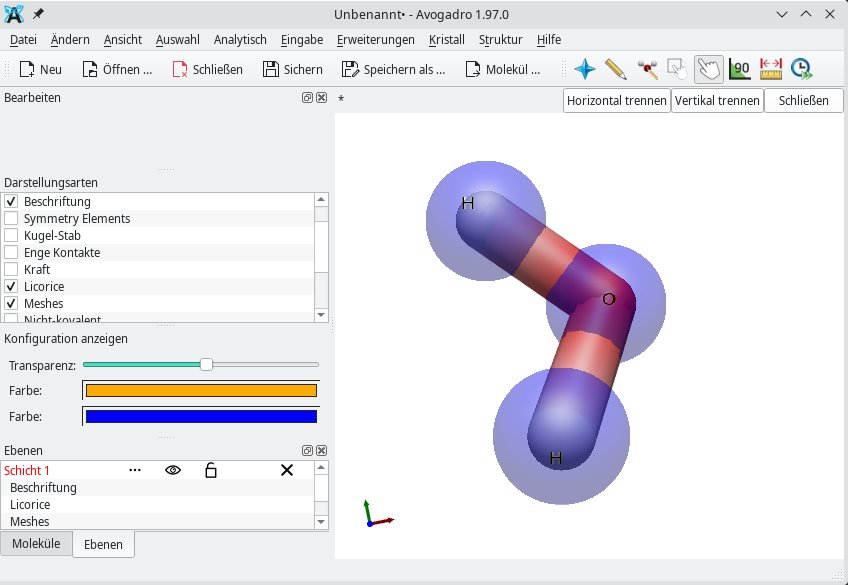
Abbildung 4: Für das Drehen von Molekülen nutzen Sie am besten die Darstellungsart Licorice.
Generell ist das Zoomen der einzelnen Moleküle nicht von der Darstellungsart abhängig, Sie aktivieren es schlicht per Mausklick auf das Auswahlwerkzeug. Bei gedrückter linker Maustaste zoomen Sie beliebig in das Molekül oder heraus. Das Verschieben des Moleküls erfolgt im selben Modus, jedoch bei gedrückter rechter Maustaste. Darüber hinaus können Sie Atome markieren, indem Sie mit der linken Maustaste darauf klicken. Ein Druck auf [Entf] löscht das Atom anschließend. Alternativ ließe es sich mittels des Zeichenwerkzeugs entfernen, indem Sie mit der rechten Maustaste auf das entsprechende Atom klicken.
Zu den anderen Aktionen, die Sie auf markierte Atome anwenden können, gehören das Anpassen der Bindungslänge und das Verschieben des markierten Atoms. Dazu aktivieren Sie zunächst das Manipulationswerkzeug und passen das Objekt anschließend bei gedrückter linker Maustaste an (Abbildung 5). Sobald Sie das Molekül bewegen, ist es sehr wahrscheinlich nicht mehr entlang den Achsen ausgerichtet. Sie schaffen Abhilfe, indem Sie Ansicht | Align View to Axes aufrufen, was die Anordnung des Moleküls animiert.
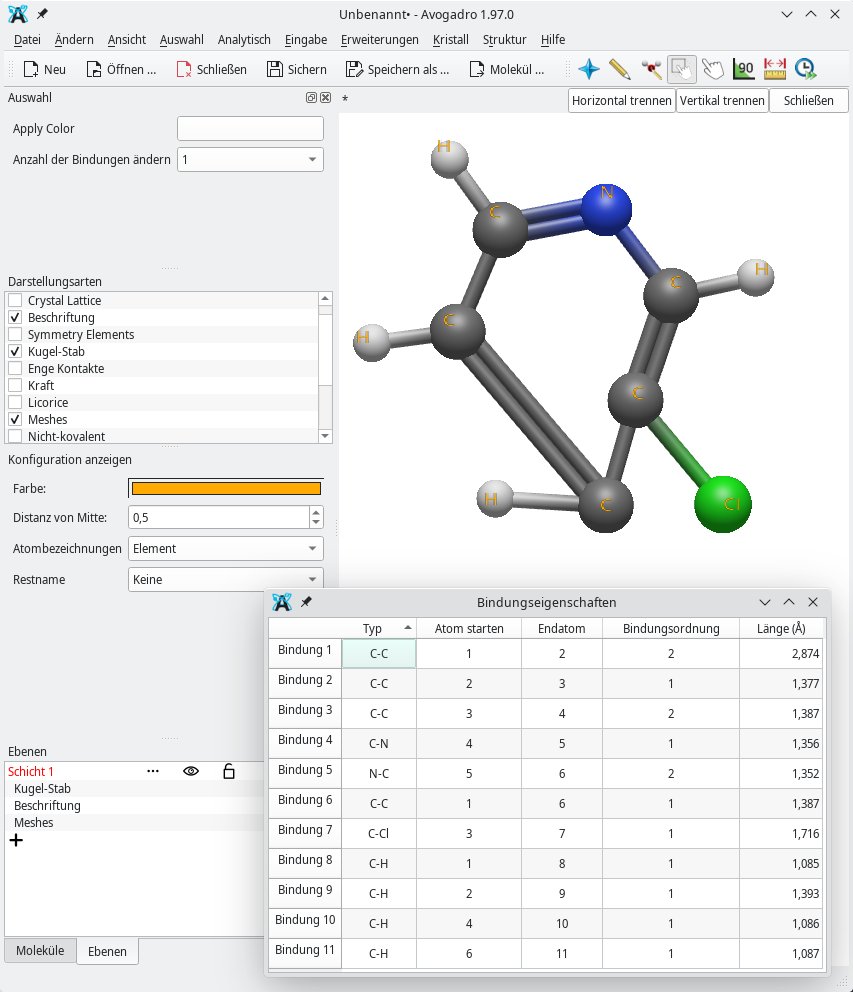
Abbildung 5: Aufgrund der Verschiebung des untersten Kohlenstoffatoms ist diese C-C-Bindung am längsten.
Moleküleigenschaften
Moleküle verfügen über naturwissenschaftliche Eigenschaften, die mit Messungen in Zusammenhang stehen. Hier ist es ratsam, mithilfe des Darstellungsmodus Beschriftung die Bezeichnung der einzelnen Atome einzublenden. Das vereinfacht das Beurteilen der Eigenschaften.
Die Eigenschaften eines Moleküls finden sich vor allem unter Analytisch | Eigenschaften. Beispielsweise informiert das Untermenü Molekülorbital über das Gewicht, die Zahl der Atome sowie die Summenformel (Abbildung 6). Üblicherweise ordnet Avogadro 2 jedem Atom eine bestimmte Farbe zu. Falls Sie die konkrete Farbzuordnung nicht kennen, schlagen Sie sie über das Untermenü Atomeigenschaften nach oder verwenden dazu die Periodentafel unter Erweiterungen | Periodensystem.
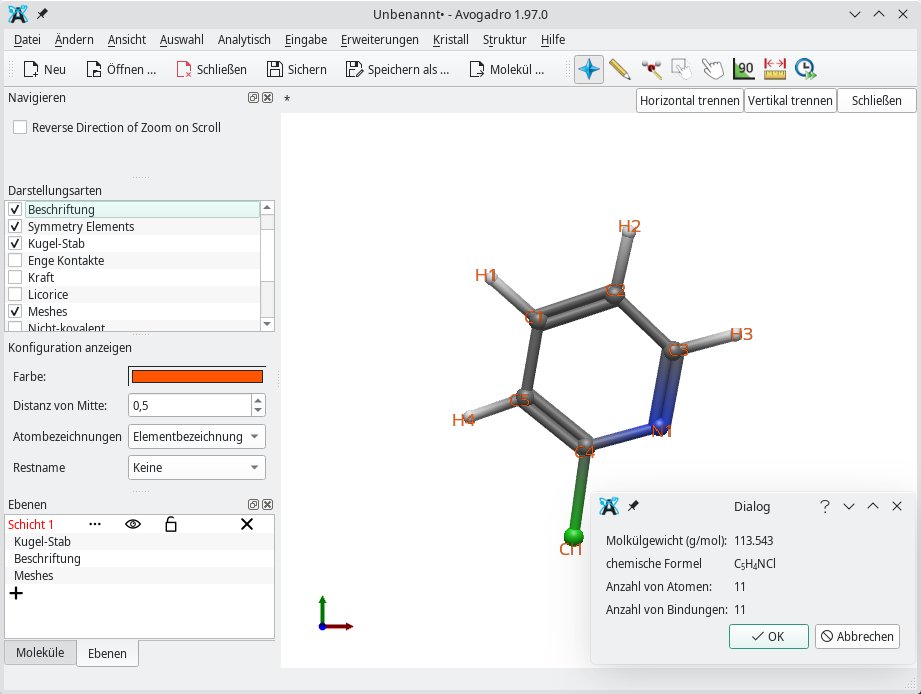
Abbildung 6: Avogadro 2 kann anhand einer Zeichnung das Molekülgewicht berechnen.
Wissenswertes liefert zudem das Untermenü Bindungseigenschaften. Hier spielen vor allem die Bindungslängen zwischen den einzelnen Atomen und die dazugehörige Bindungsordnung (siehe Tabelle “Bindungsordnungen”) eine Rolle. Sie können so zum Beispiel unter Einbeziehung der Entfernung zwischen zwei miteinander verknüpften Atomen auf die Größe des Dipolmoments schließen. Die Bindungsordnung verrät die Stärke einer Bindung. Darüber hinaus ist es wichtig, die Winkel [8] zwischen zwei Atomen oder Elektronenpaaren zu kennen. Sie lassen sich durch Aufrufen des Untermenüs Winkeleigenschaften ermitteln. Passend dazu liefert das Untermenü Torsionseigenschaften die Torsionswinkel [9].
|
Ordnung |
Art |
|---|---|
|
1 |
Einfachbindung |
|
2 |
Doppelbindung |
|
3 |
Dreifachbindung |
Ob ein Molekül ausgewogene Proportionen um eine Ebene oder Achse aufweist, lässt sich anhand der Molekülsymmetrie beschreiben, die Sie über das Untermenü Symmetrie einsehen. Ebenso bedeutend ist die üblicherweise in Ångström gemessene Distanz zwischen zwei oder mehreren Atomen, die Sie mithilfe des Messwerkzeugs (Lineal-Icon) ermitteln. Dazu markieren Sie die einzubeziehenden Atome, woraufhin Avogadro 2 links unten die Entfernung einblendet (Abbildung 7).
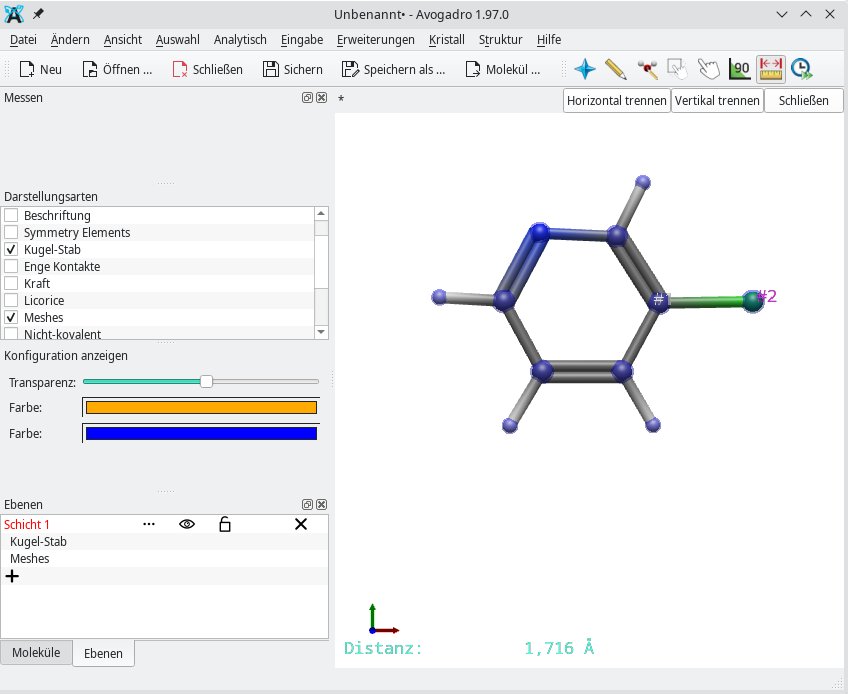
Abbildung 7: Die Entfernung zwischen zwei oder mehreren Atomen lässt sich mittels des Messwerkzeugs berechnen.
Kristalle
Kristalle dienen dazu, die Strukturen von Festkörpern zu veranschaulichen. Ein Kristall besteht aus Elementarzellen, die veranschaulichen, wie Atome, Moleküle oder Anionen räumlich zueinander angeordnet sind. Das spielt vor allem in der Halbleiterindustrie eine große Rolle, wo es unter anderem darum geht, so viele Siliziumatome wie möglich innerhalb eines winzigen Raums unterzubringen.
Zum Zeichnen von Kristallen eignet sich Avogadro 2 bestens. Im Beispiel aus Abbildung 8 kommt die einfache Verbindung Cäsiumchlorid (CsCl) mit der Koordinatenzahl 8 zum Einsatz. Letztere steht für die Zahl der Verbindungen, die vom Cäsium-Atom ausgehen. Außerdem verdeutlicht die Koordinatenzahl, wie viele Atome in eine Elementarzelle hineinpassen.
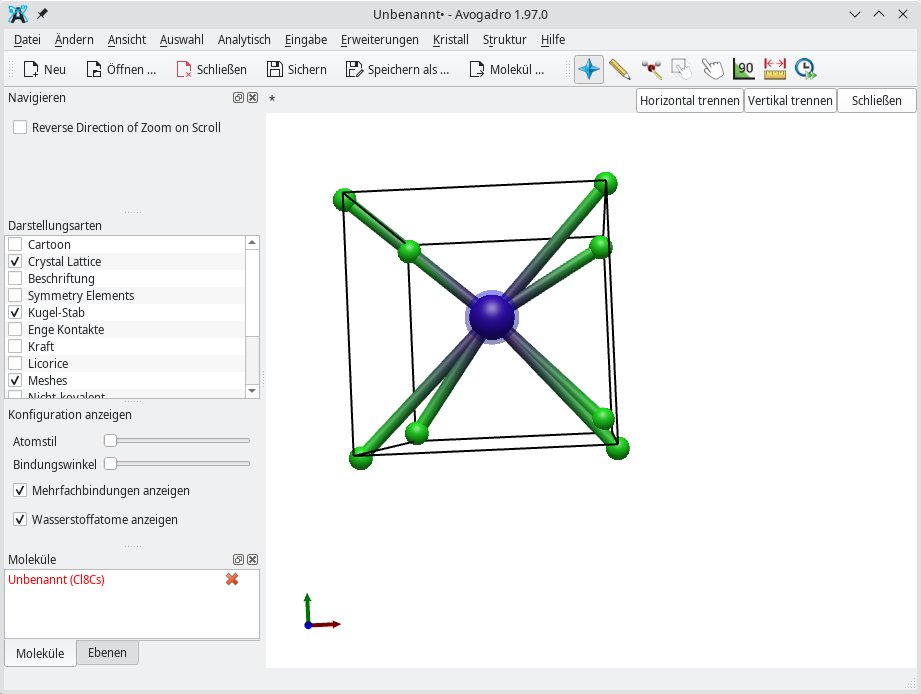
Abbildung 8: Avogadro 2 beherrscht das Zeichnen von Kristallen, wie hier der Elementarzelle für Cäsiumchlorid.
Unter Avogadro 2 benötigen Sie für Kristalle die Darstellungsarten Crystal Lattice (Kristallgitter), Kugel-Stab und Meshes. Anschließend fügen Sie eine Elementarzelle ein, die Sie unter Kristall | Elementarzelle hinzufügen finden. Der Blickwinkel auf die Elementarzelle lässt sich, genauso wie bei den Atomen beschrieben, mittels des Manipulationswerkzeugs ändern. Das Zentralatom und die anzubringenden Atome binden Sie mithilfe des Zeichenwerkzeugs ein. Es empfiehlt sich, mit dem Zentralatom – im Beispiel von CsCl ist das Cäsium – zu beginnen.
Nach Auswahl des Cäsiumatoms auf der linken Seite platzieren Sie das Zentralatom per Mausklick in der Mitte der Elementarzelle. Die mit dem Zentralatom verknüpften Chloratome setzen Sie auf die Kanten der Elementarzelle. Zunächst wählen Sie links das Chloratom aus und ziehen bei gedrückter linker Maustaste eine Linie vom Zentralatom hin zu den einzelnen Kanten des Würfels. Sobald Sie die Maustaste loslassen, fügt Avogadro 2 das Chloratom samt Bindung ein.
Achten Sie darauf, die Option Wasserstoffatome adjustieren zu deaktivieren, sodass eine einfache Bindungsordnung ausreicht. Empfinden Sie das Zentralatom im Vergleich zu den anderen Atomen als zu wuchtig, heben Sie die Skalierung der Atome auf, indem Sie nach einem Klick auf die Darstellungsart Kugel-Stab den Schalter bei Atomstil nach links verschieben.
Fazit
Die freie Software Avogadro 2 ist im Open-Source-Bereich einmalig, was sie zur Nummer eins unter den fortgeschrittenen Moleküleditoren macht. Sie bringt zahlreiche Vorlagen mit und kann gültige SMILES-Strings korrekt darstellen. Die Zeichnungen lassen sich anschließend ins SVG- und PNG-Format exportieren.
Allerdings weist die Anwendung durchaus auch Schattenseiten auf. Unerfahrene Nutzer dürften sie zunächst als wenig intuitiv zu bedienen empfinden, was die Lektüre des eingangs erwähnten E-Books erzwingt. Ein weiteres Manko liegt in der Instabilität des Programms, die vor allem bei Animationen voll zum Tragen kommt: Abstürze sind dann keine Seltenheit. Im Test gelang es zudem nicht, eine mit Avogadro 2 angefertigte Zeichnung direkt nach einem Neustart erneut zu öffnen. Dies führte lediglich zu Fehlermeldungen. (csi/jlu)
Die Autorin
Anzela Minosi bietet unter dem Pseudonym macrolab auf http://Legiit.com diverse Dienstleistungen rund um IT an. Dazu zählen Gigs, mit denen sich Daten bereinigen, auswerten sowie grafisch veranschaulichen lassen.
Glossar
- SMILES
- Simplified Molecular Input Line Entry Specification [10]. Ein proprietäres, von der Firma Daylight Inc. kontrolliertes Format für die Repräsentation eines Moleküls als Zeichenkette.
- InChI
- IUPAC International Chemical Identifier [11]. Eine Repräsentation des Moleküls als Zeichenkette. Im Gegensatz zu SMILES steht InChI als freies Format unter der LGPL.
- Van-der-Waals-Kraft
- Nicht kovalente Kräfte, die zwischen den Molekülen auftreten [12].
- Dipol
- Bei polar kovalenten Bindungen werden die Elektronen einer Bindung nicht gleichmäßig zwischen den beiden Atomen geteilt. Stattdessen zieht das elektronegativere Atom die meisten der Bindungselektronen vom weniger elektronegativeren Atom an.
- Ångström
- Ein zehnmillionstel Millimeter (10-10Meter). Keine SI-Einheit, nach DIN 1301-3 nicht mehr zugelassen. Trotzdem nach wie vor für Atomradien, Gitterabstände und Wellenlängen gebräuchlich.
Infos
- Avogadro 2: https://www.openchemistry.org/projects/avogadro2/
- E-Book zu Avogadro 2: https://dasher.wustl.edu/chem430/software/avogadro/learning-avogadro.pdf
- Avogadro-2-Appimage: https://two.avogadro.cc
- PubChem: https://pubchem.ncbi.nlm.nih.gov
- JChemPaint: Anzela Minosi, “Moleküleditor”, LU 09/2022, S. 54, https://www.linux-community.de/47870
- Molinspiration: https://www.molinspiration.com/cgi-bin/properties
- Chemical Identifier Resolver: https://cactus.nci.nih.gov/chemical/structure
- Bindungswinkel: https://de.wikipedia.org/wiki/Bindungswinkel
- Torsionswinkel: https://www.chemie.de/lexikon/Diederwinkel.html
- SMILES: https://de.wikipedia.org/wiki/Simplified_Molecular_Input_Line_Entry_Specification
- InChI: https://de.wikipedia.org/wiki/International_Chemical_Identifier
- Van der Waals: https://www.uni-due.de/~hc0014/S+WM/Definitionen/Vander.htm




